小麦全基因组关联分析工具
本平台专为小麦基因组研究打造,提供先进的全基因组关联分析(GWAS)功能,帮助研究人员高效发现与性状相关的关键基因。
通过整合多种生物信息学工具和数据资源,用户可以轻松上传基因型数据、设置分析参数,并获得详细的关联分析结果,助力育种和生物研究。
使用示例:基于全基因组关联分析(GWAS)识别小麦抗病关键位点
黄锈病是全球范围内危害最严重的小麦病害之一,常在低温高湿的气候条件下爆发,能够迅速传播并导致大面积减产,严重时损失可达70%以上。由于其病原小种演化迅速、致病谱不断变化,常规防治手段面临较大挑战。开发并应用具有广谱、持久抗性的抗病基因是实现绿色、可持续小麦生产的核心手段。
全基因组关联分析(Genome-wide association study, GWAS)是一种高效的基因挖掘策略,广泛应用于复杂性状的遗传基础解析。本研究以多个小麦自然群体为材料,利用高质量表型数据与高密度 SNP 芯片,对抗黄锈病表型进行 GWAS 分析,识别出多个与抗性显著相关的基因位点。
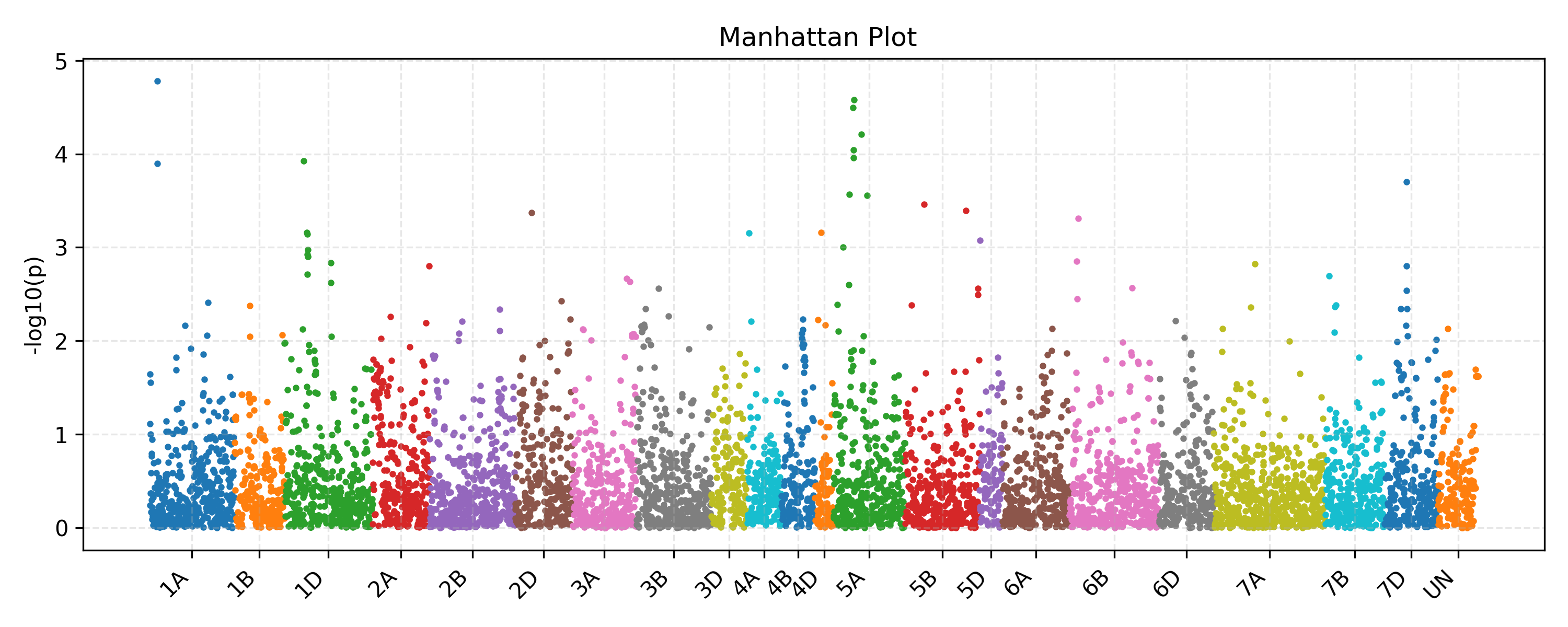
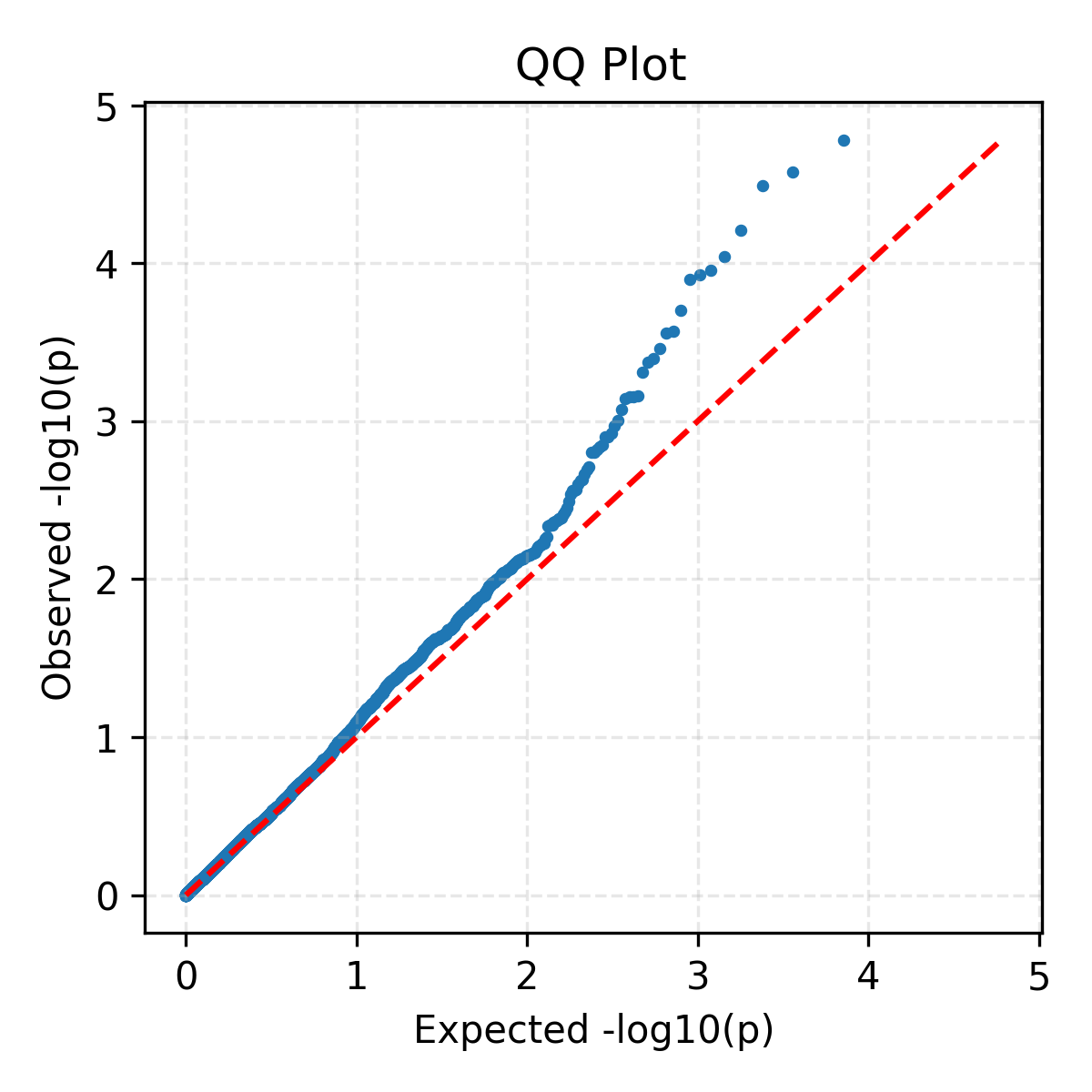
GWAS 结果采用 Manhattan 图直观展示显著 SNP 分布,同时结合 Q-Q 图验证模型合理性,并在染色体上定位候选区域,筛选潜在抗病基因。
GWAS 信号图展示
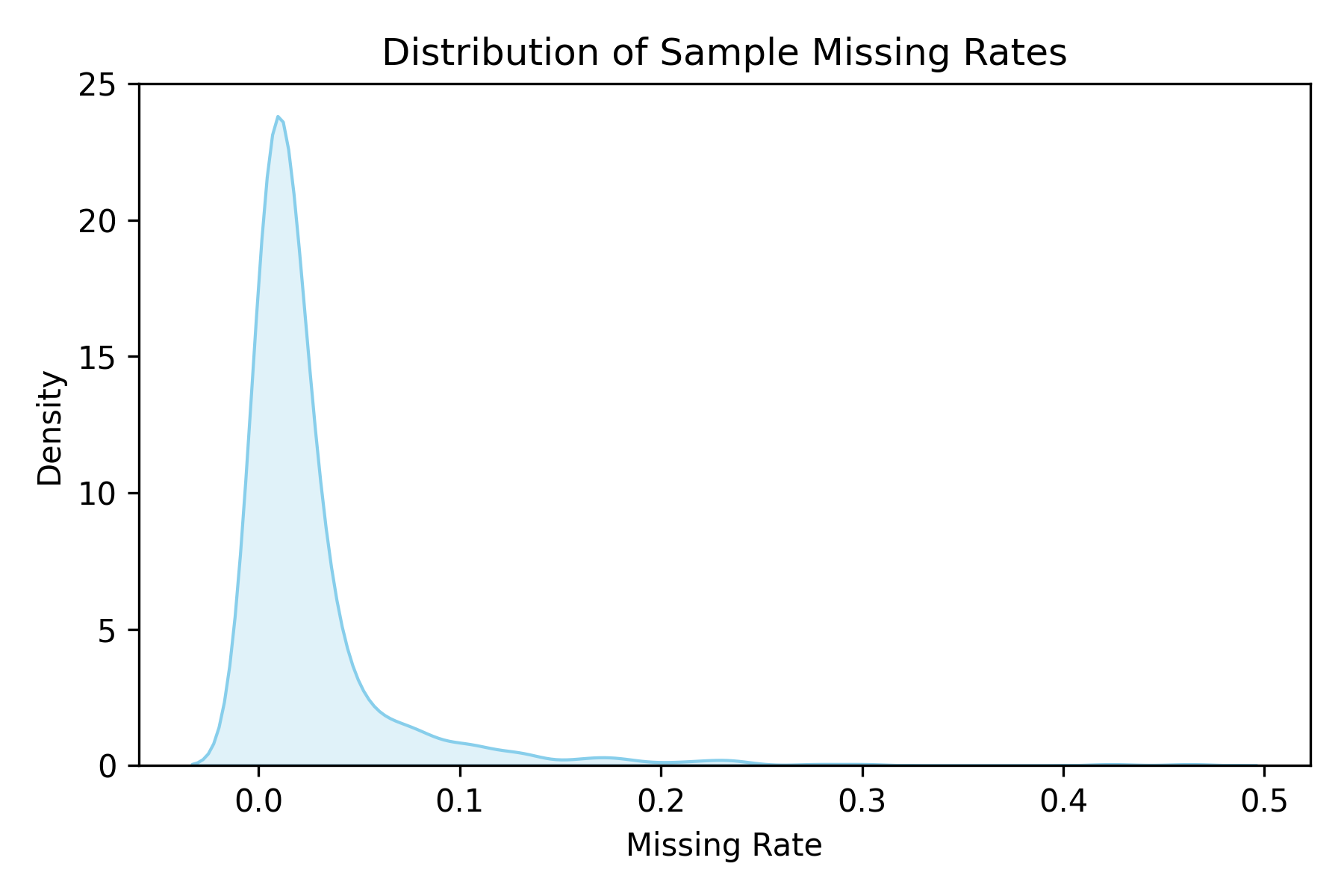
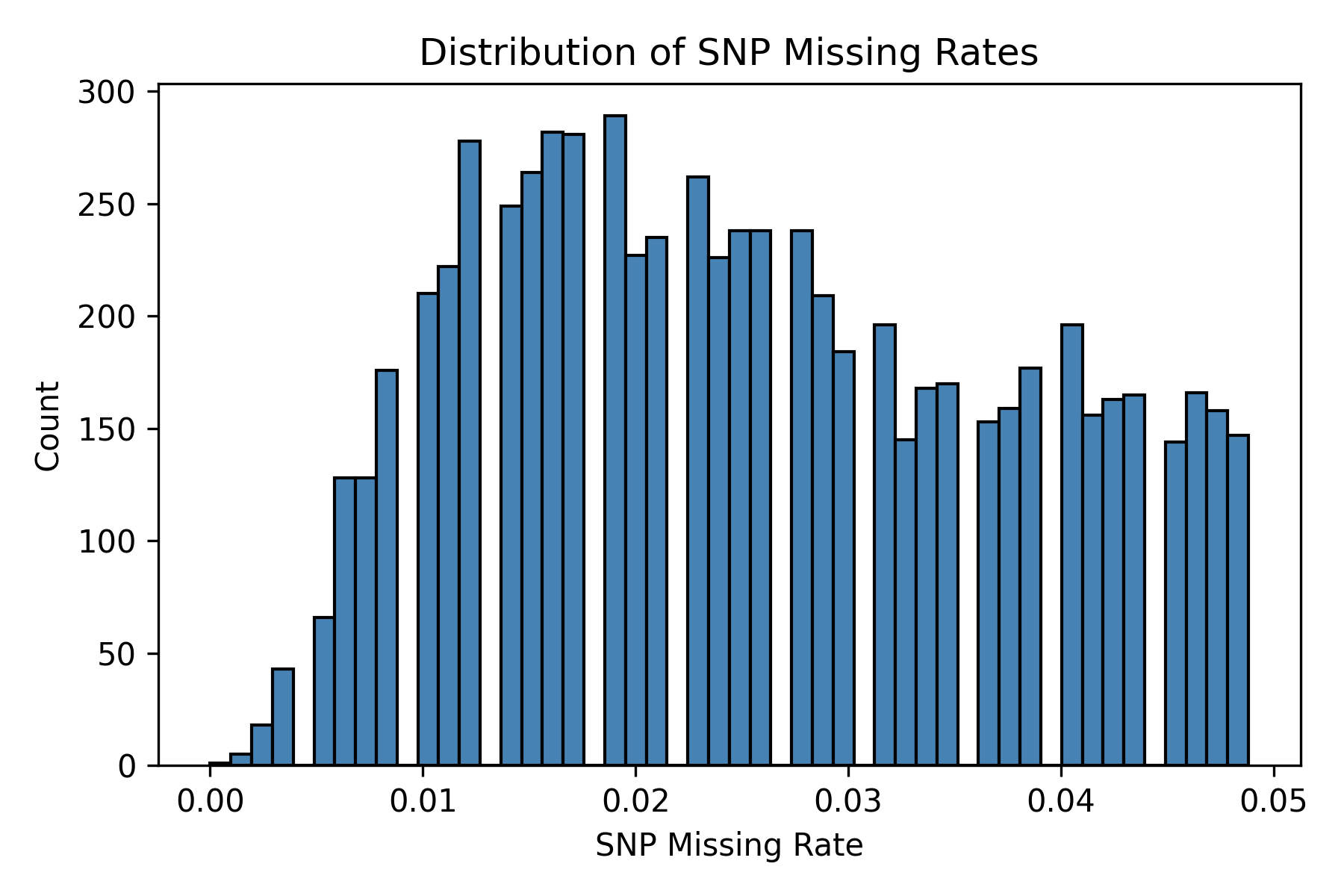
基因数据质控
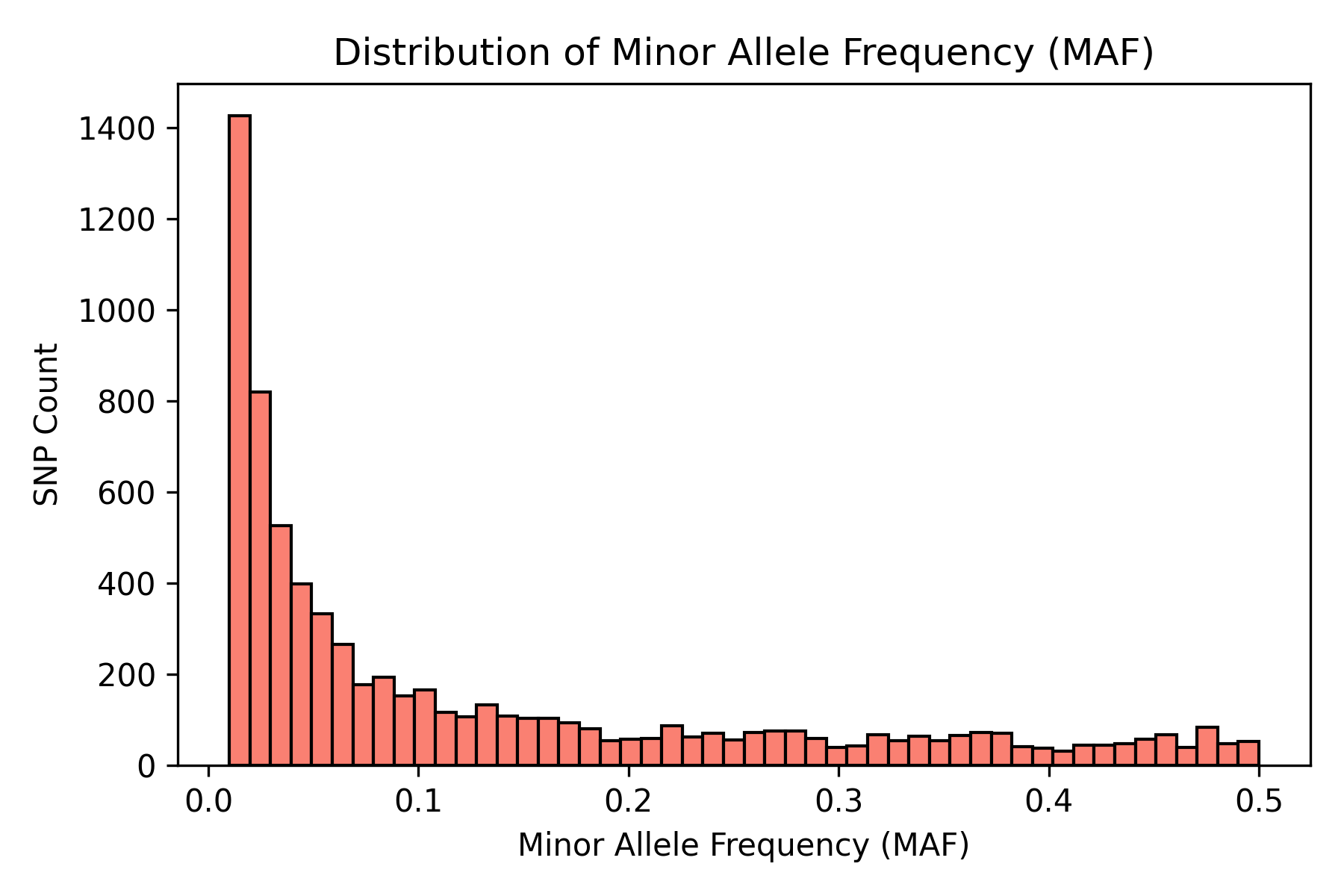
在样本缺失率密度分布图中,大多数样本的缺失率接近于 0,集中在 0-0.05 区间,呈偏态分布,仅有极少数样本缺失率超过 0.1,说明整体样本质量较高。SNP 缺失率分布相对均匀,主要集中在 0.01-0.04 区间,极少数位点缺失率过高或接近于 0,表明数据已完成初步清洗。小等位基因频率(MAF)分布呈明显右偏,大量 SNP 的 MAF 较低,其中约半数位点的 MAF 小于 0.05,提示后续关联分析中需注意低频变异的处理与阈值设定,以降低假阳性风险。
表型数据质控
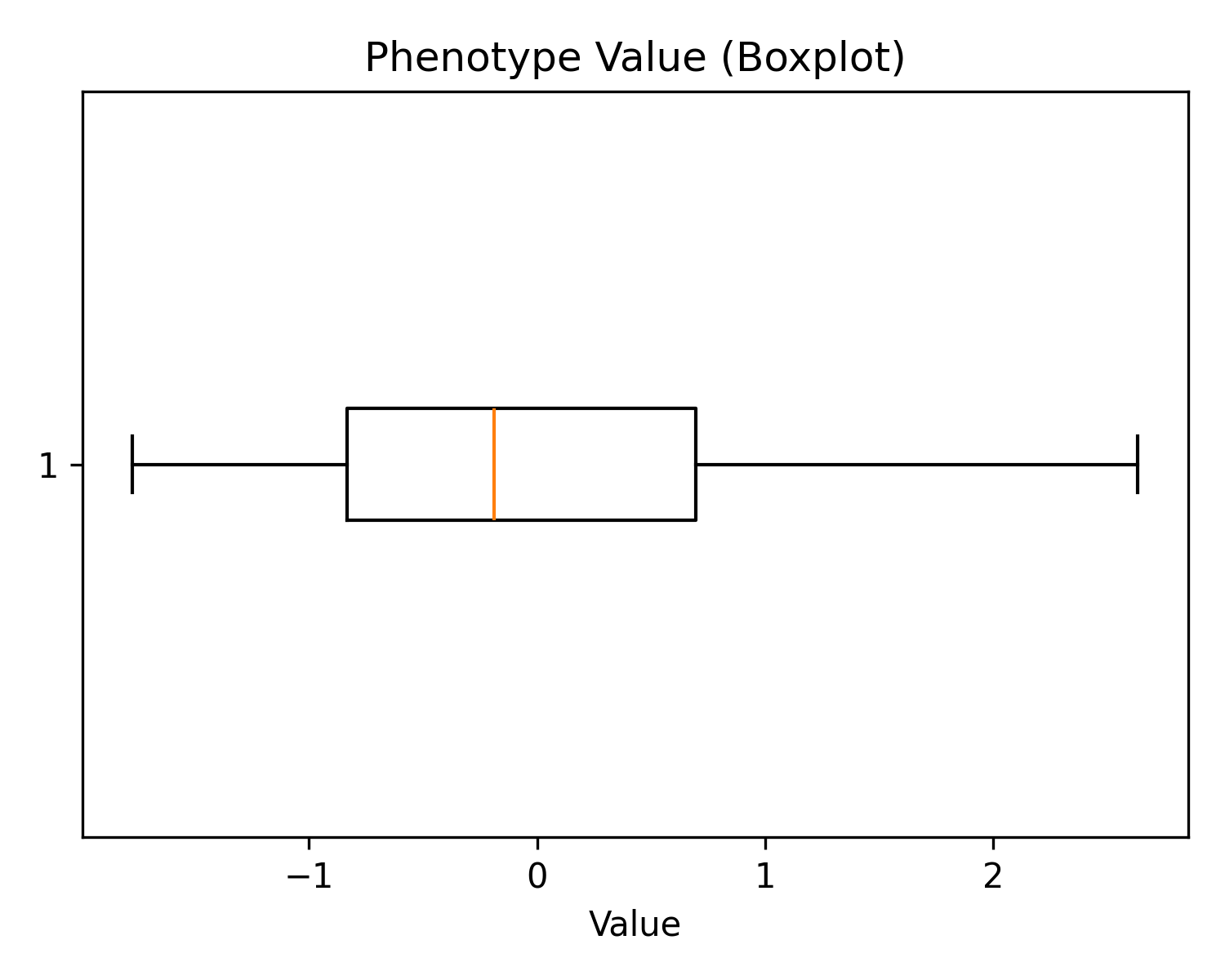
表型数据的标准化结果显示,该性状在标准化后均值约为 0(mean = 1.2e-11),标准差为 1,说明数据已经完成标准正态变换。
从分布上看,最大值约为 2.64,最小值约为 -1.77,存在一定偏斜。中位数(50%分位)为 -0.19,略低于
0,显示数据整体略偏左。
四分位间距(IQR)从 -0.83 到 0.70,说明大部分样本表型值处于该范围内。通过箱线图可以直观展示异常值的分布,有助于后续异常检测与过滤。
显著位点注释(Top 10 SNPs)
- 1. chr1A:14468537
- 2. chr5A:38144867
- 3. chr5A:35259641
- 4. chr5A:109288089
- 5. chr5A:37980147
- 6. chr5A:35532803
- 7. chr1D:18240202
- 8. chr1A:14468560
- 9. chr7D:34297040
- 10. chr5A:30026082
应用前景
基于本次 GWAS 分析结果,未来可从以下三个方面推动抗病育种的实践应用:
- 分子标记开发与早期选育: 基于显著关联的 SNP 位点构建高效分子标记,辅助抗病材料的筛选与田间抗性验证,提高选育效率。
- 候选基因功能验证与编辑: 对定位区域内的候选基因进行深入功能研究,结合转基因或基因编辑技术验证其抗病机制,加速关键基因的精确利用。
- 多基因整合与精准改良: 联合 GWAS 与结构预测等多维数据,构建抗病基因位点图谱,指导优异等位基因在不同遗传背景中的聚合与改良,推动抗黄锈病小麦品种的精准设计与选育。
上述分析结果可通过
http://gwas.seed.cesi.site/gwas_gemma/
在线访问与复现分析流程。